Lysosomal Storage Disorders Support Society
Gaucher Disease Support in India
Helping patients and caregivers in India access treatment, expert guidance, and long-term support since 2010.
What is Gaucher Disease?
Gaucher disease is a rare genetic disorder in which the body cannot properly break down a fatty substance called glucocerebroside.
This happens due to a deficiency of an enzyme called glucocerebrosidase. As a result, this fatty substance accumulates inside cells (called Gaucher cells), especially in the:
- Spleen
- Liver
- Bone marrow
This buildup can lead to symptoms such as:
- Enlarged spleen and liver
- Fatigue and anaemia
- Low platelet count (easy bruising)
- Bone pain and fractures
In India, Gaucher disease is often underdiagnosed or diagnosed late, as symptoms may be mistaken for other conditions.
Gaucher Disease in India
Gaucher disease is a rare condition worldwide, with an estimated incidence of approximately 1 in 40,000 to 60,000 births globally.
In India, the true prevalence is difficult to estimate due to underdiagnosis and limited awareness. Many cases are often misdiagnosed or detected late, as symptoms can resemble more common conditions.
However, increasing awareness and improved diagnostic facilities are helping identify more patients across the country. In some regions, a higher number of cases may be seen due to genetic factors, including consanguinity (marriage within close relatives), which can increase the risk of inherited conditions like Gaucher disease.
This highlights the importance of genetic awareness and early screening in at-risk populations.
⭐ DID YOU KNOW? The LSDSS currently supports nearly 200 registered Gaucher families across India, reflecting both the growing recognition of the disease and the need for ongoing treatment and support.
Early diagnosis and access to appropriate treatment are critical to improving outcomes for patients with Gaucher disease in India.
Video Credit: Osmosis
What Causes Gaucher Disease?
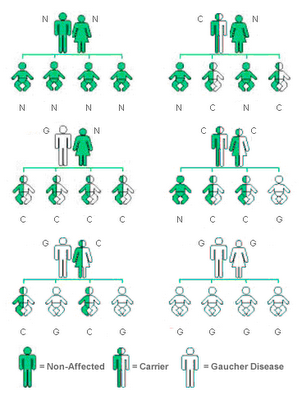
Gaucher disease is caused by mutations in the GBA1 gene, which is responsible for producing the glucocerebrosidase enzyme.
It is inherited in an autosomal recessive pattern, meaning both parents must carry the gene mutation for a child to be affected.
When this enzyme does not function properly, fatty substances accumulate in organs and tissues, leading to disease symptoms.
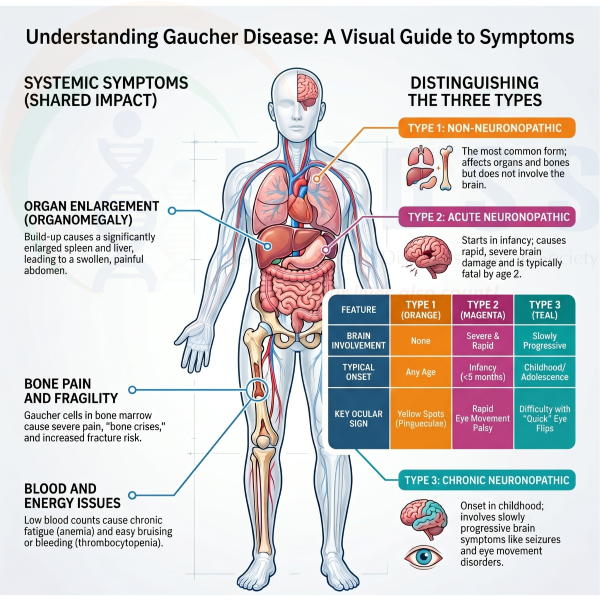
What are the symptoms of Gaucher disease?
Symptoms can vary from person to person, but common signs include:
- Enlarged spleen (abdominal swelling)
- Enlarged liver
- Fatigue and weakness (due to anaemia)
- Easy bruising or bleeding (low platelets)
- Bone pain and frequent fractures
- Delayed growth in children
⚠️ Important: These symptoms are often confused with other conditions in India, leading to delayed diagnosis.
When Should You Seek Medical Advice?
Consult a doctor if you notice:
- Persistent enlarged spleen
- Unexplained anaemia
- Frequent bone pain
- Easy bruising
Early diagnosis can significantly improve long-term outcomes.
Living with Gaucher Disease in India
Managing Gaucher disease requires long-term care, including regular monitoring, treatment, and specialist consultations.
In India, many patients face challenges such as:
- High treatment costs
- Limited access to specialised centres
- Lack of awareness
LSDSS supports patients and caregivers by helping them:
- Connect with expert doctors and treatment centres
- Access enzyme replacement therapy (ERT) programs
- Stay updated on government and support initiatives
- Join a community of patients and caregivers
👉 You are not alone — we are here to support you.
Living with Gaucher Disease?
Register with LSDSS to get treatment guidance, support, and updates.
How is Gaucher disease diagnosed?
Diagnosis usually involves:
- Enzyme test (to measure glucocerebrosidase levels)
- Genetic testing (GBA1 gene confirmation)
- Imaging tests (MRI or CT scans to assess organs and bones)
- Blood tests
In India, diagnosis may be delayed as symptoms are often mistaken for other conditions.
Early testing at specialised centres is important for timely treatment and better outcomes.
Treatment for Gaucher disease in India
There is no cure for Gaucher disease, but effective treatments are available that can significantly improve quality of life.
- Enzyme Replacement Therapy (ERT)
This is the standard and most widely used treatment.
- Given as an intravenous (IV) infusion every 2 weeks
- Replaces the missing enzyme in the body
- Helps break down accumulated fatty substances
Benefits:
- Reduces liver and spleen size
- Improves blood counts
- Reduces bone complications
Common ERT medicines:
- Cerezyme® (imiglucerase)
- VPRIV® (velaglucerase alfa)
- Elelyso® (taliglucerase alfa)
👉 ERT is considered the gold standard treatment for Gaucher disease. Although ERT is highly effective for non-neuronopathic (Type 1) Gaucher disease, it does not significantly improve neurological symptoms in Types 2 and 3.
- Substrate Reduction Therapy (SRT)
An oral (tablet) treatment option for some patients.
- Works by reducing the production of the fatty substance
- Helps decrease buildup in the body
Examples:
- Cerdelga® (eliglustat)
- Zavesca® (miglustat)
👉 Unlike ERT, SRT reduces the workload on the body rather than replacing the enzyme
- Supportive & Long-Term Care
Depending on symptoms, patients may also need:
- Blood transfusions (for severe anaemia)
- Pain management for bone disease
- Regular monitoring (MRI, blood tests)
- Physiotherapy and supportive care
Accessing Treatment in India (NPRD & Support Programs)
Accessing treatment for Gaucher disease in India can be challenging due to high costs. However, there are support mechanisms available:
Government Support (NPRD)
Under the National Policy for Rare Diseases (NPRD), eligible patients can receive financial assistance of up to ₹50 lakh for treatment at designated Centres of Excellence (CoEs).
These centres are specialised government hospitals that provide diagnosis, treatment, and access to funding support.
👉 View list of Centres of Excellence in India
However, please note that funding may be:
- One-time
- Limited because ₹50L often not enough for lifelong ERT
- Subject to approval
Additional Support
In many cases, treatment costs may exceed the government limit. Patients may also access:
- State government schemes
- Corporate social responsibility (CSR) funding
- Crowdfunding and patient support programs
How LSDSS Helps
LSDSS supports patients in navigating these pathways by:
- Connecting patients to CoEs and specialists
- Guiding families through funding applications
- Providing updates on available support programs
👉 Register with LSDSS for assistance in accessing treatment
Need Help Managing Gaucher Disease?
From diagnosis to long-term treatment, LSDSS supports patients across India. Register with us for assistance and updates!
Frequently Asked Questions (FAQs)
Get answers to some of the frequently asked questions about Gaucher's disease!
A genetic disorder caused by deficiency of an enzyme that leads to fat accumulation in organs.
It depends on the type. With treatment, many patients live normal lives.
No, but treatments like ERT and SRT can effectively manage the disease. However, research for gene therapy that could provide a potential one-time cure for Gaucher disease is currently underway.
It is inherited — both parents must carry the gene.
According to the International Collaborative Gaucher Group (ICGG) Gaucher Registry, the mean life expectancy at birth of patients with Type 1 Gaucher has been reported as 68.2 years (63.9 years for splenectomised patients and 72.0 years for non-splenectomised patients), compared with 77.1 years in a reference population (>5000 phenotypically similar patients worldwide).
Type 2 Gaucher usually results in death within the first few years of life.
Patients with Type 3 Gaucher have a shorter life expectancy, but treatment helps some patients with relatively mild neurological involvement live into their 50s.
Treatment for Gaucher is prohibitively expensive, costing anywhere from several lakhs to a few crore rupees per year, lifelong. A few companies have been providing this treatment free of cost to some patients in India under their charitable access programmes. However, at present, getting the treatment under charitable access is very difficult as the companies are not accepting any more patients. Nevertheless, you can get yourself/the patient enrolled on the waiting list for charitable treatable for Gaucher patients maintained by the International Gaucher Alliance (IGA) by filling up this form and sending it to tanya@gaucheralliance.org, with a copy to contactus@lsdssindia.org.
In addition, please get yourself/the patient enrolled on Government of India’s digital portal for crowdfunding and CSR donations for rare diseases by meeting the doctor at your nearest COE with all medical reports for medical evaluation. You can find the details of your nearest COE here.
In addition, please register yourself with the LSDSS by completing this form to get support and the latest information about the disease.