Lysosomal Storage Disorders Support Society
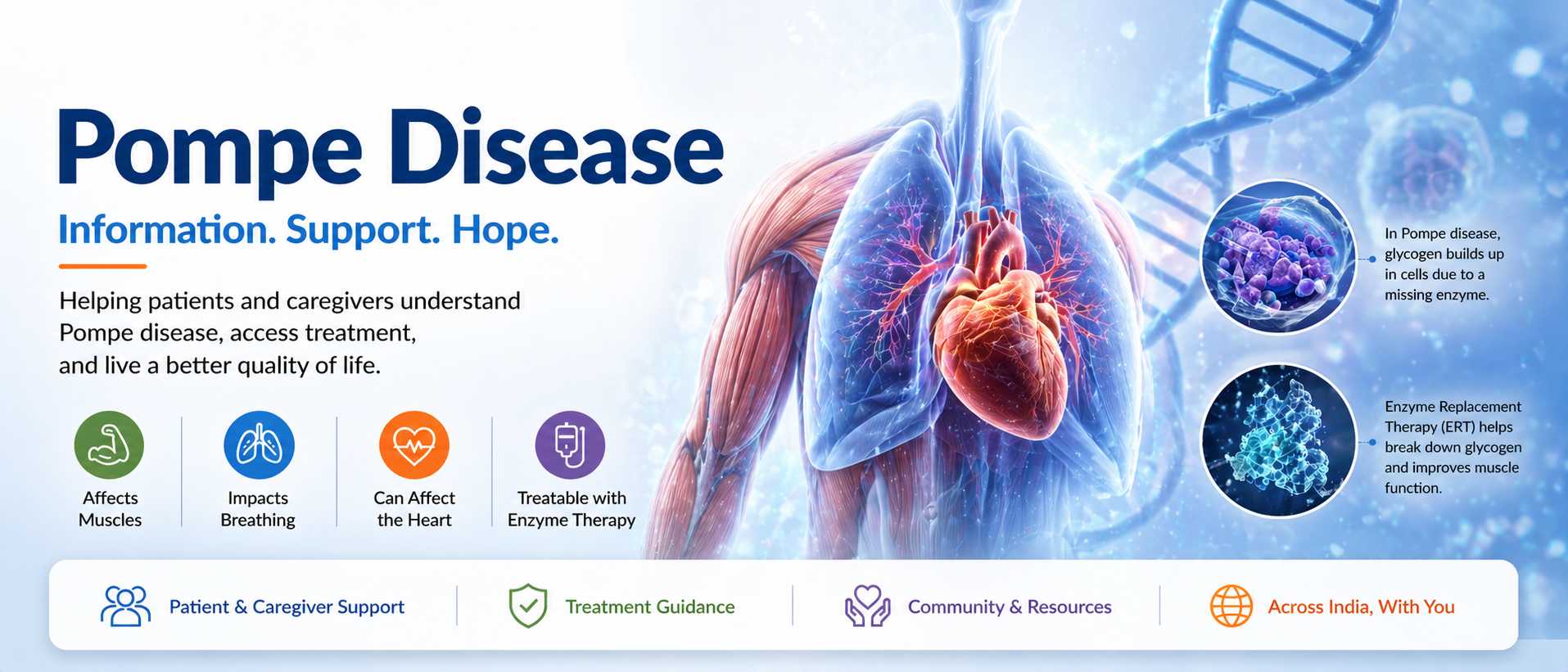
What is Pompe Disease?
Pompe disease is a rare genetic disorder caused by deficiency of the enzyme acid alpha-glucosidase (GAA), leading to accumulation of glycogen in muscles.
This primarily affects:
- Skeletal muscles
- Respiratory muscles
- Heart (in infantile form)
Over time, this leads to progressive muscle weakness and breathing difficulties.
Video Credit: Osmosis
Pompe Disease in India
Pompe disease is rare globally, with an estimated incidence of 1 in 40,000 to 300,000 births.
In India, the number of diagnosed patients is still low, largely due to underdiagnosis and lack of awareness.
However, increasing awareness and better diagnostic facilities are helping identify more patients.
LSDSS supports Pompe patients and families across India, helping them access treatment and care.
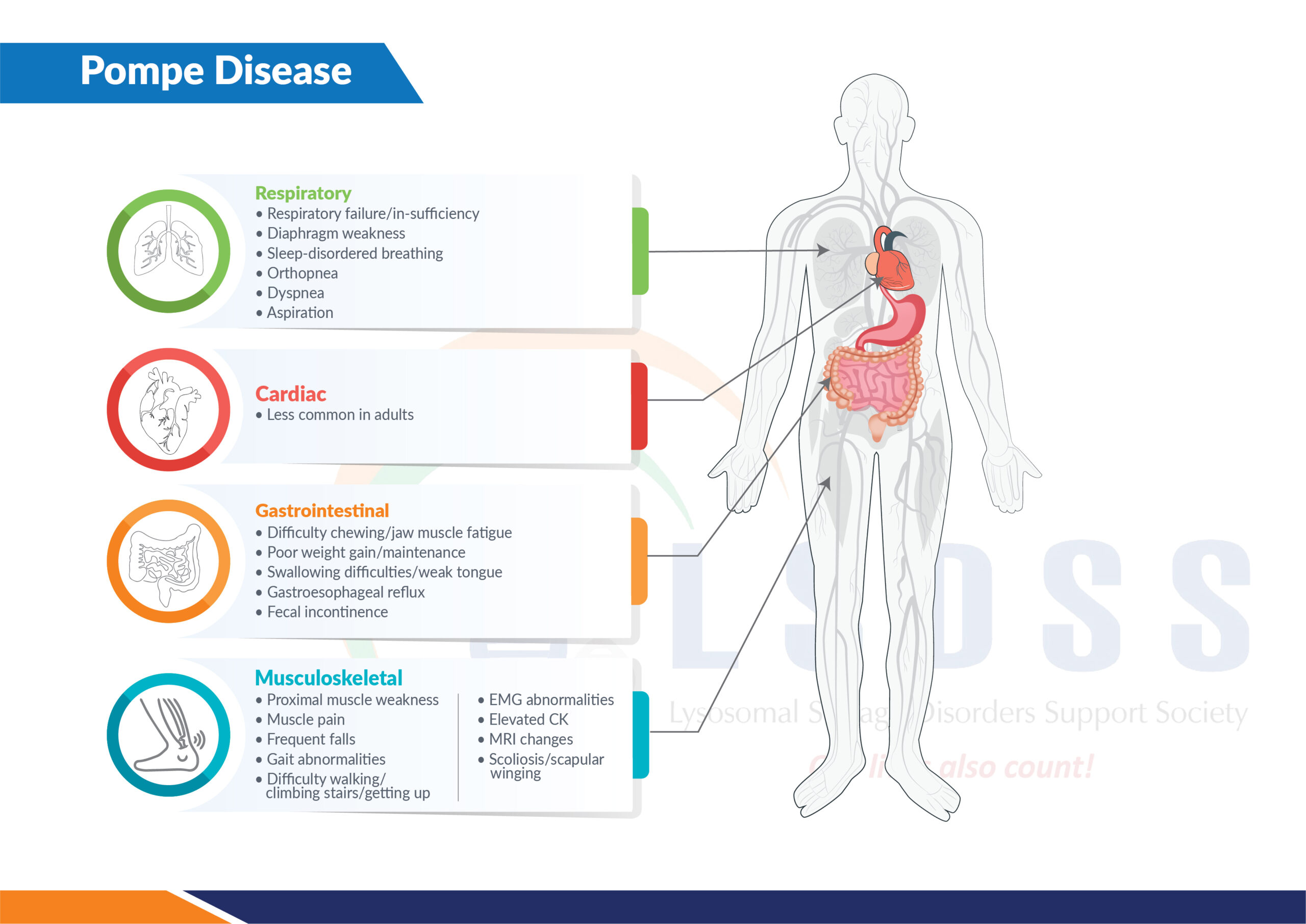
Symptoms of Pompe disease
Common symptoms include:
- Progressive muscle weakness
- Difficulty walking or climbing stairs
- Breathing problems (especially while lying down); respiratory failure is a major cause of complications
- Fatigue and reduced stamina
In infants:
- Weak muscles (floppiness)
- Enlarged heart
- Feeding difficulties
Living with Pompe Disease in India
Living with Pompe disease can be overwhelming, especially with the high cost of treatment and need for lifelong care, especially for respiratory and muscle function.
Patients often need:
- Regular monitoring
- Respiratory support
- Physiotherapy
In India, challenges include high treatment costs, delayed diagnosis, and limited access to specialised centres.
LSDSS helps patients navigate treatment, connect with experts, and access support programs.
Living with Pompe Disease?
Register with LSDSS to get treatment guidance, support, and updates.
How is Pompe disease diagnosed?
Diagnosis involves:
- Enzyme assay (GAA activity)
- Genetic testing
- Muscle biopsy (in some cases)
- Imaging and respiratory tests
Early diagnosis is critical to start treatment before irreversible damage.
Treatment for Pompe Disease
There is no cure for Pompe disease, but treatment can slow progression and improve survival.
- Enzyme Replacement Therapy (ERT)
ERT is the main treatment and replaces the missing enzyme.
- Given via IV infusion every 2 weeks
- Improves muscle function and survival
- Especially effective if started early
Common ERTs:
- Myozyme® (alglucosidase alfa)
- Lumizyme®
- Nexviazyme®
ERT improves outcomes but is not a cure and requires lifelong treatment. Moreover, it does not cross the blood-brain barrier and therefore does not significantly affect neurological involvement (if present).
2. Supportive Care
Patients may require:
- Respiratory support (BiPAP/ventilation)
- Physiotherapy
- Nutritional support
How to Access Treatment for Pompe Disease in India?
ERT is very expensive and often requires financial support and access through specialised centres.
Treatment costs can range from ₹50 lakh to over ₹2 crore per year, depending on patient weight and dosage.
Under the National Policy for Rare Diseases (NPRD), eligible patients can receive financial support of up to ₹50 lakh for treatment at designated Centres of Excellence (CoEs).
Patients must register at a CoE to access this support
👉 View list of Centres of Excellence
Additional support may include:
- State schemes
- CSR funding
- Crowdfunding
Need Help Managing Pompe Disease?
From diagnosis to long-term treatment, LSDSS supports patients across India. Register with us for assistance and updates!
Frequently Asked Questions (FAQs)
Pompe is an inherited disorder caused by the buildup of a complex sugar called glycogen in the body’s cells. The accumulation of glycogen in certain organs and tissues, especially muscles, impairs their ability to function normally.
Symptoms begin in the first months of life and include feeding problems, poor weight gain, muscle weakness, floppiness, and head lag. Respiratory difficulties are often complicated by lung infections. The heart may become enlarged (cardiomegaly), especially in infants. Many infants with Pompe also have enlarged tongues.
The classic form of infantile-onset Pompe begins within a few months of birth. Infants with this disorder typically experience muscle weakness (myopathy), poor muscle tone (hypotonia), an enlarged liver (hepatomegaly), and heart defects. Affected infants may also fail to gain weight and grow at the expected rate (failure to thrive) and have breathing problems.
The non-classic form of infantile-onset Pompe usually appears by age 1. It is characterized by delayed motor skills (such as rolling over and sitting) and progressive muscle weakness. The heart may be abnormally large (cardiomegaly), but affected individuals usually do not experience heart failure. The muscle weakness in this disorder leads to serious breathing problems,
Most individuals with late-onset Pompe experience progressive muscle weakness, especially in the legs and the trunk, including the muscles that control breathing. As the disorder progresses, breathing problems can lead to respiratory failure.
Pompe can be life-threatening if left untreated. Patients with classic infantile-onset type rarely live past 1 year of age. Patients with non-classic infantile–onset type may live to early childhood. Children with late-onset types of Pompe can live longer as the disease progresses more slowly.
Life expectancy in Pompe varies with the type of the disorder.
Classic infantile-onset Pompe is the most severe form of the disease in which symptoms appear within a few months after birth. Without treatment, affected babies often succumb to heart disease within their first year of life.
The non-classic infantile-onset form of the disease is comparatively less severe. It appears within the first year of life and has a slower progression rate, but patients often also have heart disease and breathing problems, which can be fatal if not attended to in time.
Late-onset Pompe can occur at any age. They can survive up to age 30 if the disease appears in childhood and up to age 50 if it develops in adulthood. Generally, the later the age of onset, the slower the disease progression and the longer the life expectancy.
Enzyme Replacement Therapy for Pompe can prolong survival, reverse cardiomyopathy, and improve motor function.
Patients with either type of infantile-onset Pompe may have their lives prolonged with early detection and treatment. However, if left untreated, both these types of Pompe are often fatal. Patients with classic infantile-onset type rarely live past 1 year of age. Patients with non-classic infantile-onset type may live to early childhood. Children with late-onset types of Pompe can live longer as the disease progresses more slowly.
Useful links:
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3110959/
- http://www.amda-pompe.org/
- https://medlineplus.gov/genetics/condition/pompe-disease/
- http://www.pompecenter.nl
- http://dx.doi.org/10.1038/sj.ejhg.5200367
- http://dx.doi.org/10.1086/301788
- http://dx.doi.org/10.1542/peds.2007-2222
- http://dx.doi.org/10.1002/ajmg.a.35662
- http://dx.doi.org/10.1093/hmg/3.12.2213
- http://dx.doi.org/10.1186/1750-1172-7-35
- http://dx.doi.org/10.1038/gim.2012.44