Lysosomal Storage Disorders Support Society
What is Mucopolysaccharidosis (MPS)?
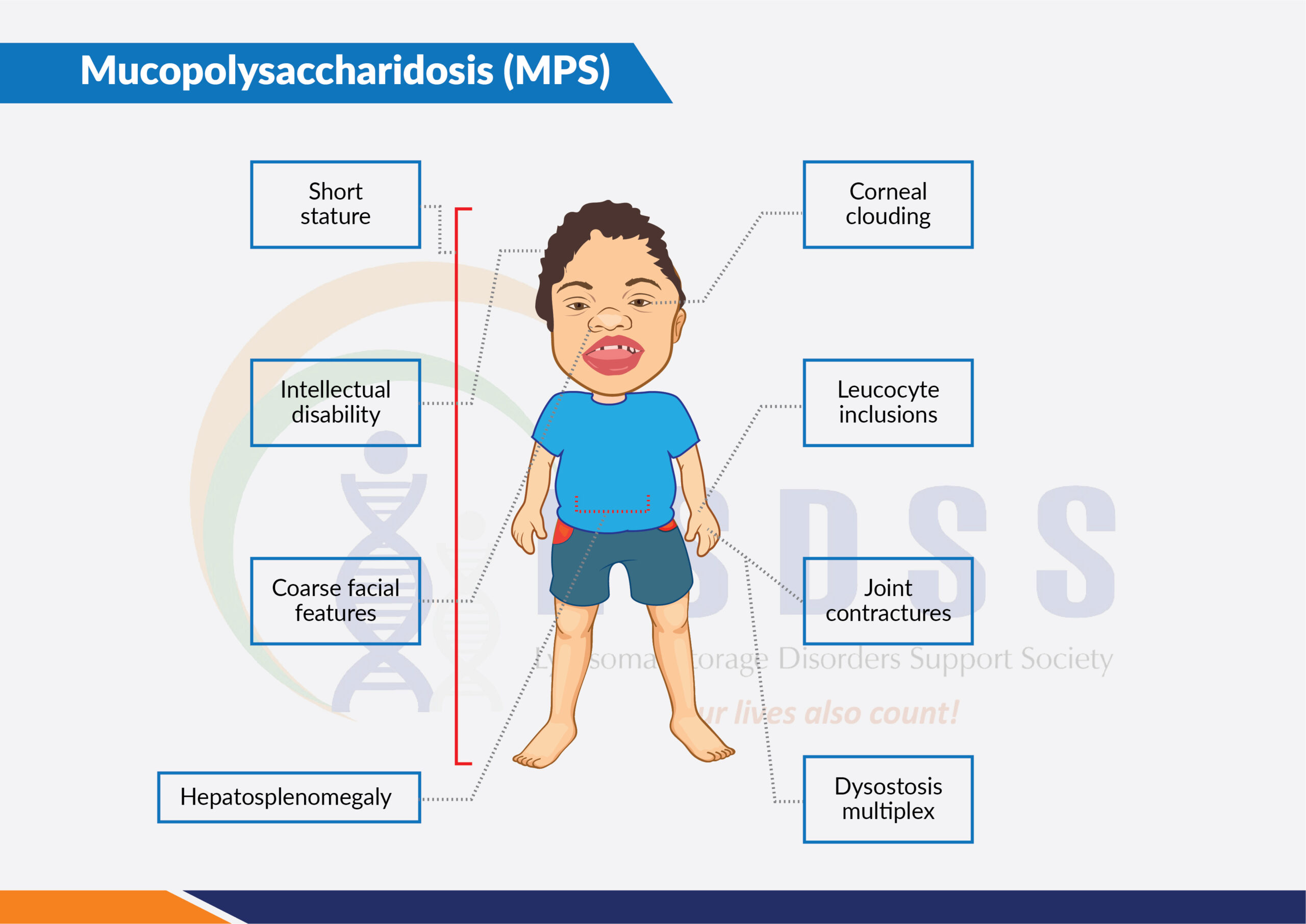
Mucopolysaccharidoses (MPS) are a group of inherited lysosomal storage disorders. Individuals with MPS disorders have a deficiency or malfunction of specific lysosomal enzymes, which leads to an abnormal accumulation of certain complex carbohydrates (mucopolysaccharides or glycosaminoglycans) in the arteries, skeleton, eyes, joints, ears, skin, and/or teeth. These accumulations may also be found in the respiratory system, liver, spleen, central nervous system, blood, and bone marrow. This accumulation eventually causes progressive damage to cells, tissues, and various organ systems of the body.
There is no cure for Mucopolysaccharidosis. However, enzyme replacement therapy (ERT) is currently available for five MPS disorders: MPS I, MPS II, MPS IVA, MPS VI, and MPS VII.
Are you an MPS patient or caregiver?
Register with us now to get support and know the latest developments about your disease!
MPS-I (MPS 1; Hurler/Hurler-Scheie/Scheie Syndrome)
Symptoms of Mucopolysaccharidosis I (MPS I)
People with mucopolysaccharidosis type I (MPS I) often develop clouding of the clear covering of the eye (cornea), which can cause significant vision loss. Affected individuals may also have hearing loss and recurrent ear infections.
Some individuals with MPS I have short stature and joint deformities (contractures) that affect mobility. Carpal tunnel syndrome develops in many children with this disorder and is characterized by numbness, tingling, and weakness in the hand and fingers. Narrowing of the spinal canal (spinal stenosis) in the neck can compress and damage the spinal cord.
Video Credit: Osmosis
While both forms of MPS I can affect many different organs and tissues, people with severe MPS I experience a decline in intellectual function and a more rapid disease progression. Developmental delay is usually present by age 1, and severely affected individuals eventually lose basic functional skills (developmentally regress).
In this form of the disorder, death typically occurs by age 10. Individuals with attenuated MPS I typically live into adulthood and may or may not have a shortened lifespan. Some people with the attenuated type have learning disabilities, while others have no intellectual impairments. Heart disease and airway obstruction are major causes of death in people with both types of MPS I.
Treatment
Management of the multiple organ system involvement in MPS I requires a multi-specialty and multi-disciplinary approach. Two treatment interventions are available to decrease the impact of the metabolic abnormalities patients with MPS I:
- Aldurazyme™ (laronidase), an enzyme replacement therapy (ERT) developed by Sanofi Genzyme, can significantly reduce liver size, increase height and weight, decrease joint restriction, lessen sleep apnea, and improve breathing in individuals with intermediate MPS I. The benefit of ERT in those with severe disease has not been assessed; however, one patient with severe mucopolysaccharidosis, who had been treated for 3 years with ERT, continued to experience decline in respiratory status, musculoskeletal and spinal involvement, and developmental skills. Because the recombinant enzyme is not thought to cross the blood-brain barrier, the best option to reduce the risk of cognitive impairment remains early HSCT.
- Successful hematopoietic stem cell transplantation (HSCT) increases survival, reduces facial coarseness and hepatosplenomegaly, improves hearing, and preserves normal heart function; however, cardiac valvular disease, skeletal manifestations and corneal clouding may continue to progress. If HSCT is accomplished before the evidence of significant developmental delay (usually under 2 years), the degree and rate of cognitive decline will likely be reduced.
- Combined ERT and HSCT is another option for individuals with severe MPS I. Providing ERT before and into the peri-HSCT period may improve the child’s existing respiratory and cardiac manifestations to a certain extent, which may reduce the risk of transplant-related complications.
- Intrathecal ERT, injecting the enzyme into the spinal fluid, is a therapy currently under investigation for the treatment of cognitive decline in severe MPS I.
MPS-II (MPS 2; Hunter Syndrome)
Mucopolysaccharidosis type II (MPS II; Hunter syndrome) is a rare X-linked recessive disease caused by deficiency of the lysosomal enzyme iduronate-2-sulphatase, leading to progressive accumulation of glycosaminoglycans in nearly all cell types, tissues and organs.
Symptoms
Clinical manifestations of mucopolysaccharidosis II include severe airway obstruction, skeletal deformities, cardiomyopathy and, in most patients, neurological decline. Death usually occurs in the second decade of life, although some patients with less severe disease have survived into their fifth or sixth decade.
Video Credit: Osmosis
Treatment
Elaprase™ (idursulfase), an enzyme replacement therapy (ERT) developed by Takeda, is available. Weekly intravenous infusions of idursulfase have been shown to improve many of the signs and symptoms and overall wellbeing in patients with MPS II. ERT with idursulfase has the potential to benefit many patients with MPS II, especially if started early in the course of the disease.
Even with ERT, patients with MPS II still require supportive symptomatic treatment from a wide range of specialists. A comprehensive initial assessment of each patient at diagnosis should therefore be undertaken, and should be followed by regular reviews. Supportive management and the anticipation of possible complications can greatly improve the quality of life of affected individuals and their families. Family members should be offered genetic counseling, and contact with other affected families, patients and support groups can be helpful.
MPS-III (MPS 3; Sanfilippo Syndrome)
Symptoms
Mucopolysaccharidosis type 3 (MPS-III; Sanfilippo syndrome) is marked by severe neurological symptoms. These include progressive dementia, aggressive behavior, hyperactivity, seizures, some deafness and loss of vision, and an inability to sleep for more than a few hours at a time. This disorder tends to have three main stages. During the first stage, early mental and motor skill development may be somewhat delayed. Affected children show a marked decline in learning between ages 2 and 6, followed by eventual loss of language skills and loss of some or all hearing.
Some children may never learn to speak. In the syndrome’s second stage, aggressive behavior, hyperactivity, profound dementia, and irregular sleep may make children difficult to manage, particularly those who retain normal physical strength. In the syndrome’s last stage, children become increasingly unsteady on their feet and most are unable to walk by age 10.
Thickened skin and mild changes in facial features, bone, and skeletal structures become noticeable with age. Growth in height usually stops by age 10. Other problems may include narrowing of the airway passage in the throat and enlargement of the tonsils and adenoids, making it difficult to eat or swallow. Recurring respiratory infections are common.
Treatment
There are currently no approved treatments for MPS III. Enzyme replacement therapy (ERT) has not been shown to be effective in MPS III. Bone marrow transplants have been tried on individuals with MPS III without positive results. Current MPS III research focuses on gene therapy, chaperone therapy and intrathecal enzyme therapy.
MPS-IV (MPS 4; Morquio Syndrome)
Mucopolysaccharidosis type 4 (MPS IV), also known as Morquio syndrome, is a progressive condition that mainly affects the skeleton. The rate at which symptoms worsen varies among affected individuals.
Symptoms
The first signs and symptoms of MPS IV usually become apparent during early childhood. Affected individuals develop various skeletal abnormalities, including short stature, knock knees, and abnormalities of the ribs, chest, spine, hips, and wrists. People with MPS IV often have joints that are loose and very flexible (hypermobile), but they may also have restricted movement in certain joints.
A characteristic feature of this condition is underdevelopment (hypoplasia) of a peg-like bone in the neck, called the odontoid process. The odontoid process helps stabilize the spinal bones in the neck (cervical vertebrae). Odontoid hypoplasia can lead to misalignment of the cervical vertebrae, which may compress and damage the spinal cord, resulting in paralysis or death.
In people with MPS IV, the clear covering of the eye (cornea) typically becomes cloudy, which can cause vision loss. Some affected individuals have recurrent ear infections and hearing loss. The airway may become narrow in some people with MPS IV, leading to frequent upper respiratory infections and short pauses in breathing during sleep (sleep apnea).
Other common features of this condition include mildly “coarse” facial features, thin tooth enamel, multiple cavities, heart valve abnormalities, a mildly enlarged liver (hepatomegaly), and a soft out-pouching around the belly-button (umbilical hernia) or lower abdomen (inguinal hernia). Unlike some other mucopolysaccharidosis types, MPS IV does not affect intelligence.
The life expectancy of individuals with MPS IV depends on the severity of the symptoms. Severely affected individuals may survive only until late childhood or adolescence. Those with milder forms of the disorder usually live into adulthood, although their life expectancy may be reduced. Spinal cord compression and airway obstruction are major causes of death in people with MPS IV.
Condition-specific height and weight curves are now available to follow patients longitudinally with Morquio syndrome. Final height is attained for most males at age 11 years and for females at age 9 years.
Treatment
Vimizim™ (elosulfase alfa), developed by BioMarin Pharmaceutical Inc., is an enzyme replacement therapy (ERT) available for MPS IVA. There is no treatment for MPS IVB.
MPS-VI (MPS 6; Maroteaux-Lamy Syndrome)
Symptoms
Children with mucopolysaccharidosis type 6 (MPS VI; Maroteaux-Lamy syndrome) usually have normal intellectual development but share many of the physical symptoms found in Hurler syndrome.
Caused by the deficiency of enzyme N-acetylgalactosamine 4-sulfatase, Maroteaux-Lamy syndrome has a variable spectrum of severe symptoms.
Neurological complications include clouded corneas, deafness, thickening of the dura (the membrane that surrounds and protects the brain and spinal cord), and pain caused by compressed or traumatized nerves and nerve roots.
Growth is normal at first but stops suddenly around age 8. By age 10, patients develop a shortened trunk, crouched stance, and restricted joint movement. In more severe cases, children also develop a protruding abdomen and forward-curving spine. Skeletal changes (particularly in the pelvic region) are progressive and limit movement. Many children also have umbilical or inguinal hernias. Nearly all children have some form of heart disease, usually involving valve dysfunction.
Treatment
Naglazyme™ (galsulfase), developed and produced by BioMarin Pharmaceutical, Inc., is an ERT for individuals with a confirmed diagnosis of mucopolysaccharidosis VI and has been approved for use in the U.S. and in many European countries.

Aarav, MPS-VI
MPS-VII (MPS 7; Sly Syndrome)
Symptoms
Mucopolysaccharidosis type VII (MPS VII; Sly syndrome), one of the least common forms of the mucopolysaccharidoses, is estimated to occur in fewer than one in 250,000 births. The disorder is caused by deficiency of the enzyme beta-glucuronidase. In its rarest form, Sly syndrome causes children to be born with hydrops fetalis, in which extreme amounts of fluid are retained in the body.
Survival is usually a few months or less. Most children with Sly syndrome are less severely affected. Neurological symptoms may include mild to moderate mental retardation by age 3, communicating hydrocephalus, nerve entrapment, corneal clouding, and some loss of peripheral and night vision. Other symptoms include short stature, some skeletal irregularities, joint stiffness and restricted movement, and umbilical and/or inguinal hernias. Some patients may have repeated bouts of pneumonia during their first years of life. Most children with Sly syndrome live into the teenage or young adult years.
Treatment
The U.S. Food and Drug Administration has approved Mepsevii, developed by Ultragenyx, as a treatment for MPS VII. In certain circumstances, MPS VII can be effectively treated by HSCT provided that the developmental and clinical status of the individual is sound at the time of HSCT.
ML-II (I-Cell Disease) and ML-III (Pseudo-Hurler Polydystrophy)
ML II and ML III are both variations of the same disease. Individuals with more severe features have ML II, while those with ML III have less severe or attenuated features. These are very rare and only two or three individuals per 1 million births are diagnosed with ML II and ML III.
Treatment
HSCT can be effective for ML II (I-cell disease).
Clinical Trials
To get information about ongoing/future clinical trials in India for mucopolysaccharidosis, please visit here.
If you or someone you know from India has been diagnosed with Mucopolysaccharidosis (MPS), please register with us by filling this form to get further information and support.
Frequently Asked Questions (FAQs)
Typical symptoms include coarse facial features (including a flat nasal bridge, thick lips, and enlarged mouth and tongue); short stature with a disproportionately short trunk (dwarfism); abnormal bone size and/or shape (dysplasia) and other skeletal irregularities; thickened skin; enlarged organs such as liver or spleen, hernias; excessive body hair; short and often claw-like hands; and progressive joint stiffness.
The mucopolysaccharidoses are a group of inherited metabolic diseases caused by the absence or malfunctioning of certain enzymes the body needs to break down molecules called glycosaminoglycans, which are long chains of sugars (carbohydrates) present in the cells.
- MPS I is caused by an absence of, or insufficient levels of, the enzyme alpha-L-iduronidase.
- MPS II (also called Hunter syndrome) is caused by lack of the enzyme iduronate sulfatase.
- MPS IIIA is caused by the missing or altered enzyme heparan N-sulfatase
- MPS IIIB is caused by the missing or deficient enzyme alpha-N-acetylglucosaminidase
- MPS IIIC results from the missing or altered enzyme acetyl-CoA:alpha-glucosaminide acetyltransferase
- MPS IIID is caused by the missing or deficient enzyme N-acetylglucosamine-6-sulfate.
- MPS IVA is caused by missing or deficient enzyme N-acetylgalactosamine 6-sulfatase.
- MPS IVB is caused by missing or deficient enzyme beta-galactosidase.
- MPS VI (also called Maroteaux-Lamy syndrome) is caused by the deficient enzyme N-acetylgalactosamine 4-sulfatase.
- MPS VII is caused by deficiency of the enzyme beta-glucuronidase.
Clinical examination and tests to detect excess excretion of mucopolysaccharides in the urine are the first steps in mucopolysaccharidosis diagnosis. Enzyme assays (testing a variety of cells or blood in culture for enzyme deficiency) are needed to provide definitive diagnosis. Prenatal diagnosis using amniocentesis and chorionic villus sampling can verify whether a fetus is affected with the disorder. Genetic counseling can help parents with a family history of MPS determine if they are carrying the mutated gene that causes the disorders.
The average life expectancy of an untreated MPS patient is 15 years.
The treatment for mucopolysaccharidosis needs to be taken lifelong and is extremely expensive, costing anywhere from several lakhs to a few crore rupees per year. A few companies have been providing mucopolysaccharidosis treatment free of cost to some patients in India under charitable access. However, at present, getting this treatment free of cost is very difficult as the companies are not accepting any more patients for charitable access. Please get yourself/the patient enrolled on the Government of India’s portal for crowdfunding and CSR donations for rare diseases by meeting the doctor at your nearest COE for medical evaluation. You can find the details of your nearest COE here. In addition, request the doctor to explore the possibility of getting charitable access to treatment by contacting these companies, and also get yourself registered with the LSDSS by completing this form.
Seven distinct mucopolysaccharidosis types and numerous subtypes have been identified. Although each mucopolysaccharidosis (MPS) differs clinically, most individuals experience a period of normal development followed by a decline in physical and/or mental function. The seven types of mucopolysaccharidosis are MPS I, MPS II, MPS III, MPS IV, MPS VI, MPS VII and MPS IX (which is extremely rare).
In Asia, most patients with MPS have MPS II (Hunter Syndrome).
Mucopolysaccharides are long chains of sugar molecules found throughout the body, often in mucus and fluid around the joints. In MPS disorders, the body is unable to properly breakdown the mucopolysaccharides, as a result of which these sugars buildup in cells, blood and connective tissue, causing a variety of health problems.
Useful links:
- https://rarediseases.org/
- http://www.clinicaltrials.gov
- http://www.mpssociety.org
- https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Mucopolysaccharidoses-Fact-Sheet
- https://omim.org/entry/253220
- http://www.geneticalliance.org/
- http://www.mpssociety.org.uk/
- http://www.ncbi.nlm.nih.gov/pubmed/4265197
- http://www.ncbi.nlm.nih.gov/pubmed/19224584
- http://www.ncbi.nlm.nih.gov/pubmed/8644704